"structRNAfinder"
| gi_545778205_gb_U000_429 | ||
cmsearch-Rfam |
||
Family :tRNA |
Id:RF00005 |
Type:Gene; tRNA; |
Taxonomy:Eukaryota; Bacteria; Viruses; Archaea; | Ontology:GO:0030533 triplet codon-amino acid adaptor activity |
 |
Description:Transfer RNA (tRNA) molecules are approximately 80 nucleotides in length. Their secondary structure includes four shortdouble-helical elements and three loops (D, anti-codon, and T loops). Further hydrogen bonds mediate the characteristic L-shaped molecular structure. tRNAs have two regions of fundamental functional importance: the anti-codon, which is responsible for specific mRNA codon recognition, and the 3' end, to which the tRNAs corresponding amino acid is attached (by aminoacyl-tRNA synthetases). tRNAs cope with the degeneracy of the genetic code in two manners: having more than one tRNA (with a specific anti-codon) for a particular amino acid; and 'wobble' base-pairing, i.e. permitting non-standard base-pairing at the 3rd anti-codon position. | ||
Score:61.2 |
E-value:8.7e-13 |
|
From sequence:4496405 |
To sequence:4496486 |
|
Alignments: NC | ||
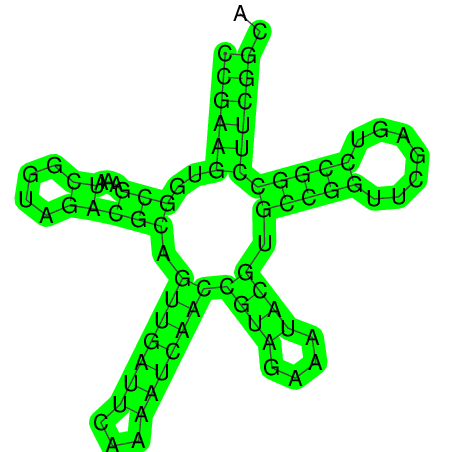
RNAfold |
||
| >gi_545778205_gb_U000_429 CCGAAGUGGCGAAAUCGGUAGACGCAGUUGAUUCAAAAUCAACCGUAGAA AUACGUGCCGGUUCGAGUCCGGCCUUCGGCA ((((((..(((...((....))))).(((((((...)))))))((((....)))).(((((.......))))))))))).. (-27.20) |
 |
|
Length sequence: 81 |
Length match:80 |
|